


日本沼虾(Macrobrachium nipponense),又称青虾、河虾,是一种重要的经济性甲壳类动物,分类学上隶属于长臂虾科(Palaemonidae)、沼虾属(Macrobrachium)。它们适应性强、分布广、食性杂、生长快、养殖效益高,因此成为中国淡水虾类养殖中的重要资源之一[1-2]。近年来,随着人们对健康饮食需求的不断增加,沼虾的市场需求也在逐渐增长。同时,科学家们也在探索如何更好地利用沼虾的营养和药用价值,为人类带来更多的健康福利。微卫星DNA(Microsatellite) 是一种由少量核苷酸(1~6个)为基本单位串联重复形成的一段序列,也被称为短串联重复序列(Short tandem repeat,STR) 或简单重复序列 (Simple sequence repeat,SSR) 。SSR多态性分析技术已被广泛应用于种群遗传结构分析、种群遗传多样性检测、遗传图谱的构建以及生产性状位点的连锁分析和QTL分析等方面[2⇓⇓⇓-6]。目前有关日本沼虾SSR分子标记开发的报道相对较少。本研究利用测序分型方法开发日本沼虾的SSR分子标记,以期为其种质选育、遗传多样性的检测等提供基础。
1 材料与方法
1.1 基因组测序文库构建
试验所用日本沼虾包括钱塘江群体(QT)、武义群体(WY)、高塘湖群体(GT)、肇庆群体(ZQ)和南宁群体(NN),均于2021年采自国内5个日本沼虾野生区域,分别采集样本6尾进行测序文库构建,取其尾部肌肉后,用无水乙醇保存备用。基因组DNA的提取采用常规的酚氯法[7]。对基因组DNA进行定量后,用Covaris M220超声波仪将DNA剪切成片段,并通过以下步骤纯化以制备测序文库[8]:对DNA片段进行末端过滤、加A,并与测序接头连接,进而进行Illumina测序。最后,构建的文库通过双末端测序过程进行测序。上海派森诺生物科技有限公司完成了测序过程。对原始读数的数据质量控制采用Fastp ver.0.20.1预处理程序[9],使用默认参数。
1.2 多态性SSR序列的筛选
1.3 SSR的测序分型
利用SSRgenotyper软件[14],对30尾日本沼虾进行了SSR的基因分型,对原程序的部分代码进行了改动,以直接读取MISA搜索的结果,修改后的代码可在 https://github.com/zergger/SSRgenotyper地址中获取。参数设置为Q=30、S=1、M=0.1、B=30、m=3,输出格式设置为genepop格式,以自行筛选的含SSR的序列作为参考序列。分型之前,利用软件BWA-MEM ver.1.0.5[15]将fastp过滤后获得的clean reads比对到包含SSR的序列上,使用软件SAMtools ver.1.6[16]删除重复、低质量的clean reads后,获得用于基因分型的SAM文件。基于获得的SAM文件,利用SSRgenotyper软件获得最终的SSR分型结果。采用TouchDown PCR,使用HotStart MasterMix(购自康为世纪生物科技股份有限公司)及5~10个样本对其中3对引物进行了PCR验证。PCR扩增条件:95 ℃预变性5 min;98 ℃10 s,退火5 s,72 ℃15 s,起始退火温度60 ℃,每个循环降低1 ℃,共10个或12个循环;98 ℃10 s,48 ℃或50 ℃退火5 s,72 ℃15 s,共30个循环;72 ℃延伸5 min,4 ℃保存。采用3%琼脂糖凝胶进行常规电泳。
1.4 数据处理
2 结果与分析
通过ebwt2InDel软件从WY_4和NN_5两个样本的测序数据中寻找包含InDel标记的序列779条。通过本实验室开发的无参考基因组SSR开发方法从779条含InDel标记的序列中筛选出含多态性SSR的序列137条,去除包含复合型SSR的序列26条,最终得到包含完美型SSR的序列111条。以包含完美型SSR的111条序列为模板,利用SSRgenotyper软件对5个群体共30个样本进行SSR分型。测序分型结果显示,111条含SSR的序列中,共有19个SSR可以成功分型,分型成功率为17.12%。分型成功的19个SSR均为3碱基重复4~6次,其中以ATC/ATG为单位的重复序列占比最多,为6个(31.58%)。利用引物设计软件Primer3针对分型成功的19个SSR序列设计了引物19对(表1)。采用TouchDown PCR对其中3对引物进行了PCR验证。由于Cluster813下游引物的GC含量少,Tm值较低,因此PCR验证中将退火温度降低至48 ℃。由图1可见,3对引物均可扩增,扩增产物大小与预期大小基本一致。
表1 测序分型法获得的日本沼虾SSR多态性位点引物的基本信息
Tab.1
| ID | 重复序列 Repeat | 上游引物(5’-3’) Forward primers | 下游引物(5’-3’) Reverse primers | 预期大小 Forecast size |
|---|---|---|---|---|
| Cluster813 | (AAT)5 | TAACTACCATGTTGTTACCGTTTAT | AAATATATAAAGACTGAATCATCAA | 91 |
| Cluster1035 | (ACA)4 | AACAACAACATCATCATCATTACTA | AAACATTACTACGACGCCTTC | 159 |
| Cluster1233 | (ACG)5 | ATCATCCTCATTCTCATTATCATC | ATTGATCGTATATACTTGATTGACG | 129 |
| Cluster1434 | (AGA)4 | ATTTATTAACGAAAGAGGAATTGAA | TTAGAGTATCTAAATATGCCACCAC | 152 |
| Cluster2134 | (ATC)4 | ATAATACAATTGATCTATATCGCGG | AACAACGAGAAATGGATTATGA | 90 |
| Cluster2253 | (ATG)4 | AGCATTCAAGAACATGGTATAAA | TGTAACAAGAATAACAACGGACTAT | 118 |
| Cluster2259 | (ATG)6 | AGTAATTTAACATGTCAGGATGAAG | CATTATGATAAATGTAACGTTTGCT | 157 |
| Cluster2265 | (ATG)5 | TTTAACAGTCGTAGTGATAATAATGG | GTCGTCCTTGTCCTATCATC | 113 |
| Cluster2269 | (ATG)4 | TATAACCTCGAGTTGAGAAA | CAACAGTAGAAAGATATGTGATTGA | 101 |
| Cluster2409 | (ATT)4 | ATGGGAATAAATAACTCTACTCGTC | TGATGATGATGATATTTATCAGTGTA | 130 |
| Cluster2414 | (ATT)4 | AGTGCATGAATGAACCTATGTCT | TATCCGTGTAATGGGAAGATTA | 155 |
| Cluster2689 | (CAA)5 | CACGTTGGTAACGTTATTATGACTA | ATTGTCGTTGTTAAATTCTTCTTCT | 138 |
| Cluster2690 | (AAC)5 | CGTAAAGGAGAATACAAAGGAGTAT | TCATTCCATATACATTAAATTGGTT | 220 |
| Cluster4103 | (CTT)4 | TTAGAGAAAGATAGAGAGAGGAAGG | ATCAGAGGGATTAACTTGTGAAC | 129 |
| Cluster4302 | (GAA)4 | AATTTCATTTAATAAACAATCGAGA | GAGTAATTTGATTTCGTCTTCTTC | 136 |
| Cluster5053 | (GCG)4 | AAGCGACAATGAATAAGATCA | ATTTGTTATACAAGTTGTTGTTGGT | 81 |
| Cluster6083 | (GTT)4 | AGGTATCGTCATTGGTTGAA | AGAAATCCAACACCTTACTAAAGAA | 211 |
| Cluster6259 | (TAA)4 | TTACACTAATCTCGTCCATTTCTT | TAAACTCACTCCTCTTAAAGACAAA | 142 |
| Cluster6879 | (TCA)4 | TAATCATGTCCTAAGATACCTCCTA | CTTCCATACAGTATTCAAAGATGAT | 154 |
图1
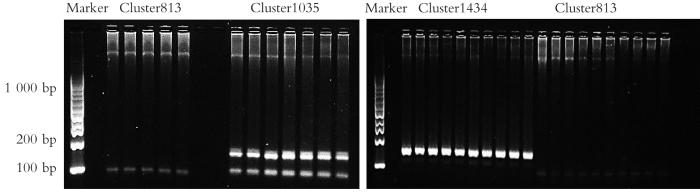
图1
选择的3对引物的PCR验证结果
Fig.1
PCR validation results of three selected primer pairs
表2 测序分型法获得的日本沼虾SSR多态性位点的特征分析
Tab.2
| ID | 重复序列 repeat | 分型成功率/% Gene type rate | Na | Ho | He | SI | PHW | PIC |
|---|---|---|---|---|---|---|---|---|
| Cluster813 | (AAT)5 | 96.67 | 2 | 0.655 | 0.448 | 0.632 | 0.002 5* | 0.344 |
| Cluster1035 | (ACA)4 | 93.33 | 2 | 0.536 | 0.399 | 0.581 | 0.021 9 | 0.315 |
| Cluster1233 | (ACG)5 | 100 | 3 | 0.733 | 0.484 | 0.725 | 0.004 7 | 0.378 |
| Cluster1434 | (AGA)4 | 100 | 2 | 0.200 | 0.183 | 0.325 | 0.455 3 | 0.164 |
| Cluster2134 | (ATC)4 | 93.33 | 2 | 0.250 | 0.456 | 0.641 | 0.015 3 | 0.348 |
| Cluster2253 | (ATG)4 | 100 | 2 | 0.767 | 0.494 | 0.679 | 0.001 1* | 0.368 |
| Cluster2259 | (ATG)6 | 93.33 | 5 | 0.500 | 0.416 | 0.819 | 0.926 3 | 0.376 |
| Cluster2265 | (ATG)5 | 100 | 2 | 0.900 | 0.503 | 0.688 | 0.000 0* | 0.372 |
| Cluster2269 | (ATG)4 | 96.67 | 3 | 0.690 | 0.518 | 0.761 | 0.184 7 | 0.396 |
| Cluster2409 | (ATT)4 | 100 | 3 | 0.333 | 0.292 | 0.534 | 0.612 1 | 0.260 |
| Cluster2414 | (ATT)4 | 100 | 2 | 0.700 | 0.481 | 0.666 | 0.006 7 | 0.361 |
| Cluster2689 | (CAA)5 | 93.33 | 3 | 0.536 | 0.408 | 0.647 | 0.154 4 | 0.334 |
| Cluster2690 | (AAC)5 | 100 | 2 | 0.800 | 0.488 | 0.673 | 0.000 0* | 0.365 |
| Cluster4103 | (CTT)4 | 100 | 2 | 0.633 | 0.440 | 0.624 | 0.003 2 | 0.339 |
| Cluster4302 | (GAA)4 | 100 | 2 | 0.233 | 0.210 | 0.360 | 0.372 6 | 0.185 |
| Cluster5053 | (GCG)4 | 100 | 2 | 0.967 | 0.508 | 0.693 | 0.000 0* | 0.375 |
| Cluster6083 | (GTT)4 | 93.33 | 2 | 0.571 | 0.499 | 0.683 | 0.429 1 | 0.370 |
| Cluster6259 | (TAA)4 | 100 | 2 | 0.900 | 0.508 | 0.693 | 0.000 0* | 0.375 |
| Cluster6879 | (TCA)4 | 100 | 2 | 0.967 | 0.508 | 0.693 | 0.000 0* | 0.375 |
注:Na为等位基因数量;Ho为观测杂合度;He为期望杂合度;SI为香农信息指数;PHW为Hardy-Weinberg平衡检验;PIC为多态性信息含量;* 表示经Bonferroni校正后偏离平衡(P<0.002 6)。
Notes:Na indicated the number of alleles; Ho indicated the observed heterozygosity; He indicated the expected heterozygosity; SI indicated the Shannon information index; PHW indicated the Hardy-Weinberg equilibrium test; PIC indicated the polymorphic information content; * indicated deviation from equilibrium after Bonferroni correction (P<0.002 6).
3 讨论
SSR在许多遗传应用中是首选的标记类型,包括基因组作图、保护遗传学、多倍体亲子鉴定、系统地理学和群体遗传学[22]。作为分子标记,SSR具有广泛的优势,包括高度多态性、共显性遗传以及在各种体外条件下的可重复性[23-24]。然而,传统的基于PCR、凝胶电泳或毛细管电泳的SSR开发方法存在劳动强度大、成本高、耗时长且效率低等问题。随着测序技术的进步和成本的下降,对整个群体进行SSR基因分型测序正在成为相对于传统的基于PCR/电泳方法的一种具有吸引力的替代方案。本研究使用SSRgenotyper从自行开发的共111个微卫星中鉴定出19个多态性SSR位点,基因分型效率超过90%(超过90%的个体都获得了分型结果)。
为验证SSR位点的多态性,本研究从5个地理群体中选择了30个样本,以确保验证样本的多样性;设计了19对引物来扩增筛选出19个多态性SSR位点,并对其中的3对引物进行了PCR和电泳分析。电泳结果验证了基于测序方法进行基因分型的准确性,凝胶电泳得到的扩增产物大小与预期大小基本一致。实际上,基于测序的SSR基因分型方法早已有之。在鱼类验证中,基于测序的基因分型方法的准确率超过90%[10]。Lewis D H等[14]将其应用在植物上,验证结果显示二核苷酸和四核苷酸SSR的不一致性最高(分别为5.7%和5.0%),重复次数超过15次的SSR最不一致(20.3%),但它们仅占总SSR数的不到0.4%。三核苷酸SSR是最常见的类型,占所有基因分型SSR位点的72.9%。Han J等[25]进行了基因组测序分型中SNP和SSR的比较研究,结果表明在群体遗传分析中,这两种方法具有高度一致性。此外,基于SSR的遗传分析还揭示了一些与SNP分析不同的结果[25]。
SSR核心序列的突变率相对较高(10-5~10-3),这会导致SSR长度的变化,从而产生SSR多态性[26]。SSR寡核苷酸的重复数在同一物种的不同基因型间差别很大,一般很少用于多态性分析。根据Weber J L[27]的研究,只有当双碱基重复序列的重复次数大于10次时,SSR标记才有可能表现出较高的PIC值。当重复次数大于16时,可提供的PIC值在0.5以上。这也是本实验中设置MISA搜索条件的依据:2个碱基重复6次及6次以上,3~6个碱基重复4次及4次以上。基于无参考基因组SSR方法筛选并经测序分型验证后获得的SSR标记都是三碱基重复,其重复次数都在4~6次之间,没有检测到具有多态性的双碱基重复,这可能与分型方法有关,也可能与样本或物种有关。而Lewis D H等[14]在植物上验证的结果也表明,三核苷酸SSR是最常见的类型。此外,在日本沼虾中,使用无参考基因组SSR筛选方法获得的多态性SSR标记的有效率较低,这也可能与物种有关。在三角鲂(Megalobrama terminalis)中,采用无参考基因组SSR筛选方法筛选的SSR标记中,多态性标记的比例可以达到90%以上[10]。将验证多态性的样本限定到WY和NN两个群体上,筛选出的多态性SSR标记数量可以提升到27个(文中未列出)。这表明作为参考的包含SSR的序列保守性不强造成比对率低,或者是由日本沼虾样本的序列多样性较高造成比对率低。
一些研究表明,重复次数较少的SSR标记往往难以检测出多态性。Valdes A M等[28]的研究结果显示,人类(Homo sapiens)中重复次数低于5的SSR,几乎检测不出多态性。而Smulders M J M等[29]则认为,重复次数多的SSR既能在种间又能在种内产生多态性,但重复次数少的SSR仅能在种间产生多态性。Xu Z等[30]对斑节对虾(Penaeus monodon)进行的研究也表明,核心序列重复次数较少的SSR标记往往是单态性的,或等位基因数目非常少,PIC值也偏低。因此,传统实验中设计SSR标记往往需要仔细选择重复次数较多的核心序列,以提高SSR标记的多态性和可靠性。但随着测序技术的发展,SSR基因分型测序正在成为相对于传统的基于PCR/电泳方法的一种具有吸引力的替代方案。由于SSR基因分型测序的高分辨率,即使传统实验中难以检测到的低重复次数SSR标记,其多态性也可检测到。本实验所获得的SSR序列重复数大部分在4~5次,但使用SSR基因分型测序依然可以检测到多态性,PIC值大多达到了中度信息含量,这对遗传多样性分析也是有利的。
参考文献
Introduction to microsatellites: basics, trends and highlights
[M]//
海南省6个养殖罗非鱼群体遗传差异的微卫星分析
[J].
利用筛选的20对微卫星引物对海南省6个养殖罗非鱼群体进行遗传多样性研究,分析各群体内的遗传变异和各群体间的遗传关系,比较不同群体的等位基因数、有效等位基因数、观测杂合度、期望杂合度、Shannon’s 多态性指数、平均多态性信息含量等遗传参数,并利用遗传距离构建6个罗非鱼群体的系统聚类图。结果表明:1)6个罗非鱼群体等位基因数为68个,平均等位基因数为1.992 2~2.255 8,平均观测杂合度为0.5725~0.7450,平均期望杂合度为0.469 2~0.532 3, Shannon’s多样性指数为0.719 3~0.848 6,多态信息含量为0.25<PIC<0.5,其中莫桑比克罗非鱼的遗传多样性指数最高,奥尼罗非鱼最低。2)采用邻接法(NJ)对6个罗非鱼群体进行聚类分析,表明6个罗非鱼群体分为2大支,吉富罗非鱼、尼罗罗非鱼、红罗非鱼、莫桑比克罗非鱼聚为一大支,其中吉富罗非鱼和尼罗罗非鱼聚集为一小支,红罗非鱼与莫桑比克罗非鱼聚为一小支;奥尼罗非鱼和泰奥罗非鱼聚为一支。综上所述,海南省6个养殖罗非鱼群体的遗传多样性较高,但不同群体之间存在差异;奥尼罗非鱼与泰奥罗非鱼亲缘关系较近,红罗非鱼与莫桑比克罗非鱼亲缘关系较近。
东海带鱼(Trichiurus japanicus)肝脏转录组SSR和SNP特征分析
[J].本研究以东海带鱼转录组测序数据为基础,采用MISA软件进行SSR筛选,利用Samtools和GATK进行SNP筛选,并进行SSR和SNP的特征分析。结果共获得SSR 49311个,其中完整的微卫星共有44625个,复合型SSR数目为4687个。SSR片段长度11~374bp。重复长度小于20bp的序列,有30997条,其中长度在12~20bp的SSR为7523个,约占总数的29.35%,呈中等多态性,重复长度大于20bp长序列为13627个,约占总数的53.18%,呈较高多态性;共获得SNP 位点数876199 个,其中颠换335218个,转换540981个,转换、颠换比例为1.61:1。在非编码区的SNP 数量为578020个;在编码区的SNP总数298179个,在编码区的碱基转换类型中C->T占比最高,为22.09%。另本研究提供了SSR和SNP原始引物各10对。研究结果可为今后带鱼的分子标记开发、遗传连锁图谱构建以及种质资源保护等提供基础数据。
Isolation of high-molecular-weight DNA from suspension cultures of mammalian cells using proteinase K and phenol
[J].
A large genome center’s improvements to the Illumina sequencing system
[J].
Fastp: an ultra-fast all-in-one FASTQ preprocessor
[J].Quality control and preprocessing of FASTQ files are essential to providing clean data for downstream analysis. Traditionally, a different tool is used for each operation, such as quality control, adapter trimming and quality filtering. These tools are often insufficiently fast as most are developed using high-level programming languages (e.g. Python and Java) and provide limited multi-threading support. Reading and loading data multiple times also renders preprocessing slow and I/O inefficient.
Pipeline for developing polymorphic microsatellites in species without reference genomes
[J].Microsatellites, also known as simple sequence repeats (SSRs), are the preferred type of marker for many genetic applications. In conjunction with the ongoing development of next-generation sequencing, several bioinformatic tools have been developed for identifying SSRs from genomic or transcriptomic sequences. Although these tools are handy for generating polymorphic SSRs, their application almost always depends on an existing reference genome or self-assembly of the reference genome. With this in mind, we propose a pipeline for developing polymorphic SSRs that may be applied to species without reference genomes. Using a species without a reference genome (black Amur bream; Richardson, 1846) as a model, our pipeline was able to effectively discover polymorphic SSRs. Under different R parameters of a reference-free single nucleotide polymorphisms (SNPs) caller (ebwt2InDel), a total of 258, 208, 102, and 11 polymorphic SSRs were mined. To quantify the accuracy of the polymorphic SSRs detected using our pipeline, we analyzed 25 SSRs with PCR experiments. All primers were successfully amplified, and most SSRs (23 SSRs, 92%) were polymorphic. From the 36 individual black Amur bream, we acquired an average of 3.36 alleles per locus, ranging from one to 11. This demonstrates the effectiveness of our pipeline in identifying polymorphic SSRs and designing primers for SSR genotyping. Ultimately, our pipeline can effectively mine polymorphic SSRs for species without reference genomes, complementing SSR mining approaches based on reference genomes and helping to resolve biological issues that accompany these methods.The online version contains supplementary material available at 10.1007/s13205-022-03313-0.© King Abdulaziz City for Science and Technology 2022, Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Variable-order reference-free variant discovery with the Burrows-Wheeler Transform
[J].In [Prezza et al., AMB 2019], a new reference-free and alignment-free framework for the detection of SNPs was suggested and tested. The framework, based on the Burrows-Wheeler Transform (BWT), significantly improves sensitivity and precision of previous de Bruijn graphs based tools by overcoming several of their limitations, namely: (i) the need to establish a fixed value, usually small, for the order k, (ii) the loss of important information such as k-mer coverage and adjacency of k-mers within the same read, and (iii) bad performance in repeated regions longer than k bases. The preliminary tool, however, was able to identify only SNPs and it was too slow and memory consuming due to the use of additional heavy data structures (namely, the Suffix and LCP arrays), besides the BWT.In this paper, we introduce a new algorithm and the corresponding tool ebwt2InDel that (i) extend the framework of [Prezza et al., AMB 2019] to detect also INDELs, and (ii) implements recent algorithmic findings that allow to perform the whole analysis using just the BWT, thus reducing the working space by one order of magnitude and allowing the analysis of full genomes. Finally, we describe a simple strategy for effectively parallelizing our tool for SNP detection only. On a 24-cores machine, the parallel version of our tool is one order of magnitude faster than the sequential one. The tool ebwt2InDel is available at github.com/nicolaprezza/ebwt2InDel.Results on a synthetic dataset covered at 30x (Human chromosome 1) show that our tool is indeed able to find up to 83% of the SNPs and 72% of the existing INDELs. These percentages considerably improve the 71% of SNPs and 51% of INDELs found by the state-of-the art tool based on de Bruijn graphs. We furthermore report results on larger (real) Human whole-genome sequencing experiments. Also in these cases, our tool exhibits a much higher sensitivity than the state-of-the art tool.
Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.)
[J].
Primer3—new capabilities and interfaces
[J].
SSRgenotyper: a simple sequence repeat genotyping application for whole-genome resequencing and reduced representational sequencing projects
[J].
BWA-MEME: BWA-MEM emulated with a machine learning approach
[J].The growing use of next-generation sequencing and enlarged sequencing throughput require efficient short-read alignment, where seeding is one of the major performance bottlenecks. The key challenge in the seeding phase is searching for exact matches of substrings of short reads in the reference DNA sequence. Existing algorithms, however, present limitations in performance due to their frequent memory accesses.This paper presents BWA-MEME, the first full-fledged short read alignment software that leverages learned indices for solving the exact match search problem for efficient seeding. BWA-MEME is a practical and efficient seeding algorithm based on a suffix array search algorithm that solves the challenges in utilizing learned indices for SMEM search which is extensively used in the seeding phase. Our evaluation shows that BWA-MEME achieves up to 3.45x speedup in seeding throughput over BWA-MEM2 by reducing the number of instructions by 4.60x, memory accesses by 8.77x, and LLC misses by 2.21x, while ensuring the identical SAM output to BWA-MEM2.The source code and test scripts are available for academic use at https://github.com/kaist-ina/BWA-MEME/.Supplementary data are available at Bioinformatics online.© The Author(s) (2022). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Twelve years of SAMtools and BCFtools
[J].SAMtools and BCFtools are widely used programs for processing and analysing high-throughput sequencing data. They include tools for file format conversion and manipulation, sorting, querying, statistics, variant calling, and effect analysis amongst other methods.
GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research——an update
[J].GenAlEx: Genetic Analysis in Excel is a cross-platform package for population genetic analyses that runs within Microsoft Excel. GenAlEx offers analysis of diploid codominant, haploid and binary genetic loci and DNA sequences. Both frequency-based (F-statistics, heterozygosity, HWE, population assignment, relatedness) and distance-based (AMOVA, PCoA, Mantel tests, multivariate spatial autocorrelation) analyses are provided. New features include calculation of new estimators of population structure: G'(ST), G''(ST), Jost's D(est) and F'(ST) through AMOVA, Shannon Information analysis, linkage disequilibrium analysis for biallelic data and novel heterogeneity tests for spatial autocorrelation analysis. Export to more than 30 other data formats is provided. Teaching tutorials and expanded step-by-step output options are included. The comprehensive guide has been fully revised.GenAlEx is written in VBA and provided as a Microsoft Excel Add-in (compatible with Excel 2003, 2007, 2010 on PC; Excel 2004, 2011 on Macintosh). GenAlEx, and supporting documentation and tutorials are freely available at: http://biology.anu.edu.au/GenAlEx.rod.peakall@anu.edu.au.
Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment
[J].Genotypes are frequently used to identify parentage. Such analysis is notoriously vulnerable to genotyping error, and there is ongoing debate regarding how to solve this problem. Many scientists have used the computer program CERVUS to estimate parentage, and have taken advantage of its option to allow for genotyping error. In this study, we show that the likelihood equations used by versions 1.0 and 2.0 of CERVUS to accommodate genotyping error miscalculate the probability of observing an erroneous genotype. Computer simulation and reanalysis of paternity in Rum red deer show that correcting this error increases success in paternity assignment, and that there is a clear benefit to accommodating genotyping errors when errors are present. A new version of CERVUS (3.0) implementing the corrected likelihood equations is available at http://www.fieldgenetics.com.
POPGENE 1.32, the user-friendly shareware for population genetic analysis
[EB/OL]. [
Analyzing tables of statistical tests
[J].
Construction of a genetic linkage map in man using restriction fragment length polymorphisms
[J].We describe a new basis for the construction of a genetic linkage map of the human genome. The basic principle of the mapping scheme is to develop, by recombinant DNA techniques, random single-copy DNA probes capable of detecting DNA sequence polymorphisms, when hybridized to restriction digests of an individual's DNA. Each of these probes will define a locus. Loci can be expanded or contracted to include more or less polymorphism by further application of recombinant DNA technology. Suitably polymorphic loci can be tested for linkage relationships in human pedigrees by established methods; and loci can be arranged into linkage groups to form a true genetic map of "DNA marker loci." Pedigrees in which inherited traits are known to be segregating can then be analyzed, making possible the mapping of the gene(s) responsible for the trait with respect to the DNA marker loci, without requiring direct access to a specified gene's DNA. For inherited diseases mapped in this way, linked DNA marker loci can be used predictively for genetic counseling.
A new resource for the development of SSR markers: millions of loci from a thousand plant transcriptomes
[J].
Basic concepts and methodologies of DNA marker systems in plant molecular breeding
[J].
Strategies for microsatellite isolation: a review
[J].In the last few years microsatellites have become one of the most popular molecular markers used with applications in many different fields. High polymorphism and the relative ease of scoring represent the two major features that make microsatellites of large interest for many genetic studies. The major drawback of microsatellites is that they need to be isolated de novo from species that are being examined for the first time. The aim of the present paper is to review the various methods of microsatellite isolation described in the literature with the purpose of providing useful guidelines in making appropriate choices among the large number of currently available options. In addition, we propose a fast and easy protocol which is a combination of different published methods.
Population-level genome-wide STR discovery and validation for population structure and genetic diversity assessment of Plasmodium species
[J].Short tandem repeats (STRs) are highly informative genetic markers that have been used extensively in population genetics analysis. They are an important source of genetic diversity and can also have functional impact. Despite the availability of bioinformatic methods that permit large-scale genome-wide genotyping of STRs from whole genome sequencing data, they have not previously been applied to sequencing data from large collections of malaria parasite field samples. Here, we have genotyped STRs using HipSTR in more than 3,000Plasmodium falciparumand 174Plasmodium vivaxpublished whole-genome sequence data from samples collected across the globe. High levels of noise and variability in the resultant callset necessitated the development of a novel method for quality control of STR genotype calls. A set of high-quality STR loci (6,768 fromP.falciparumand 3,496 fromP.vivax) were used to studyPlasmodiumgenetic diversity, population structures and genomic signatures of selection and these were compared to genome-wide single nucleotide polymorphism (SNP) genotyping data. In addition, the genome-wide information about genetic variation and other characteristics of STRs inP.falciparumandP.vivaxhave been available in an interactive web-based R Shiny application PlasmoSTR (https://github.com/bahlolab/PlasmoSTR).
Informativeness of human (dC-dA)n· (dG-dT)n polymorphisms
[J].Abundant human interspersed repetitive DNA sequences of the form (dC-dA)n.(dG-dT)n have been shown to exhibit length polymorphisms. Examination of over 100 human (dC-dA)n.(dG-dT)n sequences revealed that the sequences differed from each other both in numbers of repeats and in repeat sequence type. Using a set of precise classification rules, the sequences were divided into three categories: perfect repeat sequences without interruptions in the runs of CA or GT dinucleotides (64% of total), imperfect repeat sequences with one or more interruptions in the run of repeats (25%), and compound repeat sequences with adjacent tandem simple repeats of a different sequence (11%). Informativeness of (dC-dA)n.(dG-dT)n markers in the perfect sequence category was found to increase with increasing average numbers of repeats. PIC values ranged from 0 at about 10 or fewer repeats to above 0.8 for sequences with about 24 or more repeats. (dC-dA)n.(dG-dT)n polymorphisms in the imperfect sequence category showed lower informativeness than expected on the basis of the total numbers of repeats. The longest run of uninterrupted CA or GT repeats was found to be the best predictor of informativeness of (dC-dA)n.(dG-dT)n polymorphisms regardless of the repeat sequence category.
Allele frequencies at microsatellite loci: the stepwise mutation model revisited
[J].We summarize available data on the frequencies of alleles at microsatellite loci in human populations and compare observed distributions of allele frequencies to those generated by a simulation of the stepwise mutation model. We show that observed frequency distributions at 108 loci are consistent with the results of the model under the assumption that mutations cause an increase or decrease in repeat number by one and under the condition that the product Nu, where N is the effective population size and u is the mutation rate, is larger than one. We show that the variance of the distribution of allele sizes is a useful estimator of Nu and performs much better than previously suggested estimators for the stepwise mutation model. In the data, there is no correlation between the mean and variance in allele size at a locus or between the number of alleles and mean allele size, which suggests that the mutation rate at these loci is independent of allele size.
Use of short microsatellites from database sequences to generate polymorphisms among Lycopersicon esculentum cultivars and accessions of other Lycopersicon species
[J].
Identification of abundant and informative microsatellites from shrimp (Penaeus monodon) genome
[J].Microsatellites were isolated from P. monodon genomic libraries by direct sequencing of recombinant clones without probe screening. Forty-nine out of 83 clones sequenced contained 99 microsatellite arrays of three or more repeats. When five or more and ten or more repeats were considered, 28 and 14 microsatellites were detected, respectively. The 99 microsatellites were classified as perfect (75%), imperfect (6%), compound perfect (3%) and compound imperfect (16%). The abundance of di-, tri-, tetra- and hexanucleotide repeats were 67%, 20%, 9% and 3%, respectively. The dinucleotide repeats included 36 (CT)n, 31 (GT)n, 17(AT)n and 3 (CG)n. One octanucleotide repeat (ATTTATTC)5 was found within a large repeat sequence. Optimal annealing temperatures were determined for PCR using 11 primer sets encompassing 15 microsatellites. Ten primer sets provided successful amplifications with allele sizes generally ranging from 139 to 410 bp. All these primers amplified polymorphic loci with PIC values ranging from 0.63 to 0.96. Two primer sets amplified additional bands which can easily be distinguished from the bands of the main locus. Three out of 10 P. monodon microsatellites also amplified alleles in P. vannamei. The abundance and informative nature of P. monodon microsatellites and their potential for cross-species amplification make them useful for genetic studies.

{kind=link}
{kind=link}